Neuron-damaging机制发现在继承了肌萎缩性脊髓侧索硬化症小鼠模型
新的研究揭示了什么可能是一个主要neuron-damaging侮辱,发生在一种遗传形式的一场毁灭性的神经退行性疾病。这项研究,由细胞出版社出版在8月26日的《华尔街日报》神经元,描述了一个关键的机械联系突变蛋白在动物模型和疾病发病机理的肌萎缩性脊髓侧索硬化症(ALS)。
ALS是一种疾病,会攻击大脑和脊髓的神经元控制随意运动。没有治疗ALS,通常在成年人和发展特点是一个进步的瘫痪,通常会导致死亡的三到五年内诊断。ALS约10%的病例是继承和部分那些是归因于蛋白质叫做SOD1基因突变。的确切机制,链接SOD1基因突变与运动神经元退化尚未建立。
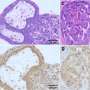
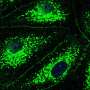
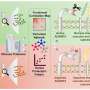
故障的线粒体,细胞内的能源生产结构,也被卷入ALS病理学。”先前的研究使用啮齿动物模型和人类样本显示,SOD1基因突变与线粒体的外表面在受影响但不影响组织,“高级研究作者解释说,唐教授w·克利夫兰从加州大学圣地亚哥。“此外,压敏电阻器阴离子通道(VDAC1),也存在于线粒体的外膜和控制线粒体和细胞的其余部分之间的通信,也与细胞死亡。”
克利夫兰教授和他的同事建立在这些早期观察和发现SOD1基因突变与VDAC1动物脊髓的表达SOD1基因突变,而这种交互VDAC1中断函数。抑制VDAC1函数是已知细胞减少能源生产和动力破坏的形成活性氧。
研究人员继续表明,突变SOD1-driven VDAC1抑制中看到脊髓线粒体突变SOD1表达动物的症状之前发达国家和在疾病进展程度增加。重要的是,减少VDAC1活动加速爆发致命的瘫痪肌萎缩性侧索硬化症小鼠。
“我们的证据表明,减少VDAC1函数和相应的减少细胞内的线粒体功能直接组件损坏从SOD1基因突变,”克利夫兰教授说。“发现VDAC1 SOD1基因突变在神经系统的目标提供了重要的见解机制根本不成熟的运动神经元的变性和死亡。”