通过筛选“垃圾”DNA可以发现致癌基因突变
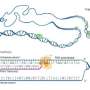
 and their target genes. Green nodes represent genes with HighD SNPs (showing high allele frequency difference among human populations) in their promoters. Size of green nodes scaled based on their degree centrality. Nodes with higher centrality are bigger and tend to be in the center. This movie shows HighD sites tend to occur in hub promoters. Credit: Vaja Liluashvili, Zeynep H. Gümüş")
研究人员现在可以识别非编码DNA中的DNA区域,非编码DNA是基因组中没有转化为蛋白质的主要部分,其中的突变可能导致癌症等疾病。
他们的方法揭示了非编码DNA中许多潜在的遗传变异发展各种不同的癌症。这种方法在发现其他致病变异方面具有巨大潜力。
与我们23,000个蛋白质编码基因所在的基因组编码区不同,构成我们基因组98%的非编码区对我们知之甚少。最近的研究强调了以前被认为是“垃圾”DNA的非编码区域在蛋白质调控中的生物学价值。这一新的信息为研究人员筛选非编码区域和识别功能最重要的区域提供了一个起点。
耶鲁大学的资深作者Mark Gerstein教授说:“我们的技术使科学家们能够专注于基因组非编码区域中功能最重要的部分。”“这不仅有利于癌症研究但也可以扩展到其他遗传疾病。”
该团队使用了1000个基因组计划第一阶段的全套遗传变异,以及ENCODE项目生成的非编码区域的信息,并确定了没有积累太多变异的区域。蛋白质编码基因在人类生存和健康中起着至关重要的作用,并且处于强大的“净化”选择之下,这消除了变异。研究小组发现,一些非编码DNA区域显示出与蛋白质编码基因几乎相同的低水平变异,并将这些区域称为“超敏感”区域。
在超敏感区域内,他们观察了特定的单个DNA字母,当这些字母被改变时,会对遗传区域造成最大的干扰。如果这个非编码的超敏感区域是许多相关基因网络的中心,变异就会引起更大的连锁反应,从而导致疾病。
他们整合了所有这些信息,开发了一种名为FunSeq的计算机工作流程。该系统根据非编码区域的遗传变异对人类疾病的预测影响来优先考虑它们。“我们的方法是一种实用而成功的方法,可以使用来自ENCODE和1000个基因组计划等免费数据,在基因组的非编码区域筛选纯化选择,”Wellcome Trust桑格研究所的作者薛亚丽博士说。“这确实显示了这些大规模开放获取数据集的价值。”
该团队将FunSeq应用于90种癌症基因组,包括乳腺癌、前列腺癌和脑肿瘤,并发现了近100种潜在的非编码癌症驱动变异。在乳房里癌症基因组例如,他们发现一个DNA字母的变化似乎对发育有很大的影响乳腺癌.这一个字母的变化发生在一个超敏感的区域,这个区域是许多相关基因网络的中心。
“虽然我们看到,第一个有效使用我们的工具是为了癌症这项研究的主要作者、威康信托基金会桑格研究所的克里斯·泰勒-史密斯博士说:“这种方法可以用于发现基因组非编码区域中任何潜在的致病变异。”“我们对这种方法的巨大潜力感到兴奋,它可以在人类这些关键但尚未开发的领域发现进一步的致病因素和有益的变异基因组."
更多信息:Ekta Khurana, Yao Fu, Vincenza Colonna, Xinmeng Jasmine Mu等(2013)。“从1092人的变异的综合注释:应用于癌症基因组学”先进的在线出版科学2013年10月3日。