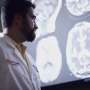
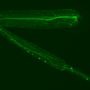
神经元的破碎机械堆积在肌萎缩性侧索硬化症
 from getting rid of defective parts. Credit: Sheng Lab, NINDS, Bethesda, MD")
健康的运动神经元需要运输损坏组件从肌肉紧张连接脊髓的胞体。如果它不能,有缺陷的组件堆积,细胞变得生病和死亡。美国国立卫生研究院的研究人员国家神经疾病和中风研究所(研究所)学习超氧化物歧化酶的基因的突变1 (SOD1),导致肌萎缩性侧索硬化症,导致细胞受损材料积累。这项研究发表在《华尔街日报》神经元表明,一个潜在的目标治疗这种家庭形式的肌萎缩性侧索硬化症。
超过12000美国人ALS,也被称为卢伽雷氏症,大约5 - 10百分比从父母那里继承了一个遗传突变。这些例家族性肌萎缩性侧索硬化症往往造成SOD1突变基因编码,一个重要的酶位于神经元的线粒体细胞的能源生产结构。这种突变导致的死亡运动神经元控制病人的肌肉,导致进行性瘫痪。
“大脑中大约90%的能量是由线粒体”Zu-Hang Sheng说,博士,一个研究所的科学家和研究的资深作者。“如果线粒体不健康,他们生产能源效率较低;他们还可以释放有害化学物质叫做活性氧导致细胞死亡。因此,线粒体损伤可引起神经退化。”
在健康的神经元,存储容器称为核内体后期收集受损的线粒体和各种破坏性的化学物质。马达叫做动力蛋白的蛋白质运输核内体结构称为溶酶体,核内体的使用化学物质分解。盛博士的研究小组发现,这种SOD1突变的神经细胞的关键过程是错误的因为SOD1基因突变干扰一个关键分子称为snapin钩子核内体动力蛋白的蛋白质。
“Snapin功能作为一个适配器连接动力蛋白的蛋白质为交通核内体,“盛博士说。“如果你块snapin函数,就会滞留在核内体和溶酶体的破坏受损的线粒体将失去他们的能力。”
盛博士和他的同事进行了实验小鼠的ALS SOD1基因突变。利用光和电子显微镜研究的co-first作者,张玉香谢,博士观察到增强受损的线粒体突变动物的运动神经纤维。这种积累在场即使在疾病的早期阶段明显症状之前出现。
“SOD1基因突变小鼠是研究模型对ALS,”盛博士说。“效果非常类似于人类发现的症状患者。”
Snapin高度动力蛋白核内体通过一个叫做动力蛋白中间的蛋白质链的一部分(DIC)。在脊髓从影响小鼠运动神经元,盛博士的研究小组发现,改变SOD1结合DIC和防止snapin这样做。增加snapin这些神经元在早期无症状的疾病的阶段纠正问题并减少了有缺陷的线粒体的形成。这帮助运动神经元生存时间,稍微增加了动物的寿命。它也减慢运动协调的损失,恶化在动物与SOD1突变运动神经元死亡。
这就解释了为什么“我们提供一种新的机械联系SOD1基因突变损害核内体运输,”盛博士说。“这可以提供细胞早期治疗干预措施的未来发展目标在运动神经元可能仍然能利用的。”
盛博士的研究是第一个在肌萎缩性侧索硬化症研究领域研究的内体交通培养运动神经元从成年小鼠模型而不是从小鼠胚胎。该研究的第一作者,Bing周,博士进行了这种技术挑战性的创新,因为成人运动神经元是一个更好的模型来研究肌萎缩性侧索硬化症等疾病症状的成年。
“通过使用成人运动神经元的老鼠模型,我们发现这个运输的缺陷,“盛博士说。”这个模型可能有用不仅为研究肌萎缩性侧索硬化症,而且其他成人神经退化疾病原因。”