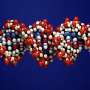
大规模基因组破坏在乳腺癌
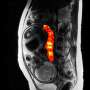
. A small segment of the gene, already massively amplified, breaks off and fuses with chromosome 8 (lower rectangle). On that chromosome, parts of the gene are copied as many as 1000 times, with various segments jumping around within the chromosome (green arcs). This shows why we want to identify HER2-positive patients as early as possible, to prevent the kind of chaos that we register here cumulatively, says Schatz. Credit: Schatz Lab, CSHL/JHU")
在癌症细胞中,基因错误造成严重破坏。拼写错误的基因,以及结构性variations-larger-scale重组DNA,可以包含大量chromosomes-disturb仔细平衡机制,它们已进化到调节细胞生长。基因通常沉默的大量激活和突变蛋白形成。这些和其他中断导致大量的问题导致细胞生长没有克制,癌症最臭名昭著的标志。
本周,冷泉港实验室的科学家们(3)发表了基因组研究的一个最详细的地图在癌症细胞的结构变化基因组。地图显示大约20000结构性变化,其中一些曾经指出由于技术限制在一个长期流行的方法基因组测序。
领导的团队,测序专家迈克尔·c·沙茨和w·理查德•McCombie读癌细胞的基因组与所谓的读测序技术。这种技术读取更长期的DNA片段比年长的短内容的技术。当结果解释与最近发表的两个复杂的软件团队,两个优势是明显的:读测序方面的丰富信息和上下文。例如,它可以更好地理解重复的DNA字母弥漫的基因组部分看到他们在一个更大的背景。
研究小组证明读技术通过使用它的力量来读的基因组细胞来源于一个叫SK-BR-3的细胞系,一个重要的模型乳腺癌细胞与HER2基因变化(有时也称为ERBB2)。大约20%的乳腺癌是“HER2阳性”,这意味着他们过度产生HER2蛋白质。这些癌症往往是最积极的。
“大多数我们确定的20000个变异细胞系中错过了短内容排序,”玛丽亚Nattestad说,博士,谁执行的工作和同事,同时仍然沙茨实验室成员3和约翰霍普金斯大学。“特别感兴趣的,我们找到了一个高度复杂的DNA变异周围的HER2基因。”
在他们的分析中,团队结合读测序的结果和结果的另一种实验,读取信息,或记录,被激活的基因生成。全面地了解了一个非常详细的描述如何扰乱癌症基因组结构变化细胞并揭示了如何癌症细胞迅速发展。
沙茨是兼职副教授3和约翰·霍普金斯大学布隆伯格杰出副教授和McCombie 3教授说这是“基本继续建立一个目录的变体癌症细胞类型使用最好的技术。读排序是一个宝贵的工具来捕获复杂的结构性变化,我们期待它的广泛采用,用于研究和临床实践中,尤其是在测序成本进一步下降。”
进一步探索