优化的基因编辑工具防止遗传性耳聋小鼠的听力损失,而没有检测到脱靶效应

哈佛医学院和波士顿儿童医院的科学家们使用了一种新的基因编辑方法来挽救患有遗传性听力损失的小鼠的听力,并成功地做到了这一点,并且没有因为治疗而产生任何明显的脱靶效应。
这些被称为贝多芬老鼠的动物接受了与人类渐进性听力丧失相同的基因突变治疗,在25岁左右达到严重耳聋。
这项新方法于7月3日在网上公布自然医学这项研究涉及经典CRISPR-Cas9基因编辑系统的优化、更精确的版本,该系统能够更好地识别贝多芬小鼠中发现的致病突变。这种改进的工具允许科学家选择性地使听力基因Tmc1的缺陷副本失效,同时保留健康的副本。
值得注意的是,研究人员报告说,他们的系统成功地在小鼠基因组的30亿个字母中识别出了一个不正确的DNA字母。
研究人员警告说,即使是这样一种高度精确的基因编辑疗法也要在人类身上应用,还有很多工作要做。然而,他们表示,这项工作是一个里程碑,因为它极大地提高了标准基因编辑技术的有效性和安全性。
“我们的研究结果表明,这种现在经典的CRISPR/Cas9编辑工具的更精细、更有针对性的版本达到了前所未有的识别和准确性,”该研究的联合高级研究员、哈佛医学院布拉瓦尼克研究所Bertarelli转化医学教授David Corey说。
此外,研究小组表示,该结果为使用相同的精确方法治疗其他由基因的单个缺陷副本引起的主要遗传遗传疾病奠定了基础。
每个人都继承了同一基因的两个副本——分别来自父母。在许多情况下,一个正常的基因足以确保正常的功能,使个体免于疾病。相比之下,在所谓的显性遗传疾病中,一个有缺陷的拷贝就能导致疾病。
“我们相信我们的工作为超靶向治疗一系列由基因缺陷副本引起的遗传疾病打开了一扇门,”研究联合高级研究员杰弗里·霍尔特说,他是哈佛医学院波士顿儿童医院耳鼻喉科和神经病学教授,他也隶属于波士顿儿童医院的F.M. Kirby神经生物学中心。“这真的是精准医疗。”
携带有缺陷Tmc1基因的小鼠被称为贝多芬小鼠,因为它们的疾病过程模仿了这位著名作曲家经历的渐进性听力丧失。然而,路德维希·范·贝多芬失聪的原因仍然是一个猜测的问题。
在老鼠身上,贝多芬缺陷是由Tmc1基因DNA序列中一个错误的字母(A而不是t)标记出来的,这个错误拼出了正常听力和耳聋之间的区别。
禁用或沉默Tmc1基因的突变拷贝足以保护动物的听力,但如何才能在不无意中也使健康基因失效的情况下做到这一点呢?
两把钥匙胜过一把钥匙
经典的CRISPR-Cas9基因编辑系统通过使用引导分子- grna -来识别目标突变DNA序列。一旦目标DNA被精确定位,切割酶cas9就会将其剪切。
到目前为止,这些基因编辑器显示出不太完美的准确性。这是因为引导Cas9酶到达目标位点的引导RNA和切割目标DNA的Cas9酶并不完全精确,最终可能切割错误的DNA。
为了克服这些挑战,研究人员采用了最初由麻省总医院病理学教授Keith Joung和病理学助理教授Ben kleinver开发的工具,该工具使用了源自金黄色葡萄球菌的改良Cas9酶,而不是源自化脓性链球菌的标准Cas9酶。
为了提高检测和破坏的准确性,新的优化系统结合了两个级别的识别- grna定位目标基因和Cas9的修饰形式,可以精确定位贝多芬小鼠中的特定DNA突变。使用两种形式的鉴定确保了精确和选择性地切割该基因的异常副本(且仅是异常副本)。
该研究的第一作者Bence Gyorgy说:“我们利用了这个系统识别突变DNA而不是正常DNA的事实,并使用双重识别系统来提高精度。”Bence Gyorgy在哈佛医学院期间进行了这项工作,现在在瑞士巴塞尔的分子和临床眼科研究所工作。“这种方法在靶向突变基因方面达到了前所未有的特异性。”
在最初的一组实验中,在有和没有贝多芬突变的细胞中,该工具准确地区分了Tmc1基因副本中的突变DNA和正常DNA。进一步的分析显示,在包含一个缺陷基因和一个正常基因副本的贝多芬细胞中,至少99%的分子“切割”只发生在有缺陷的基因副本中。
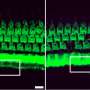
接下来,研究人员将基因编辑疗法注射到带有和没有贝多芬突变的小鼠的内耳中。DNA分析表明,编辑活动只发生在贝多芬缺陷小鼠的内耳细胞中。在没有突变的小鼠内耳细胞中没有检测到编辑变化,这一发现证实了该工具的准确性。
为了确定基因编辑疗法是否干扰了正常的基因功能,科学家们刺激了不携带贝多芬缺陷的治疗小鼠内耳的听觉细胞(称为毛细胞)。这些细胞显示出不变的、正常的听力反应,证实基因编辑疗法对正常的基因功能没有影响。
沉默贝多芬
为了衡量这种疗法是否在动物身上而不仅仅是在细胞上起作用,研究人员进行了黄金标准的听力测试。他们测量了动物的听觉脑干反应,记录了内耳毛细胞检测到多少声音并传递到大脑。
如果不进行治疗,贝多芬小鼠通常在6个月大时完全失聪。相比之下,没有这种基因缺陷的老鼠一生都能保持正常的听力,并能探测到大约30分贝的声音,这一水平与耳语相似。
在接受基因编辑治疗两个月后,贝多芬小鼠的听力明显优于未经治疗的携带基因突变的兄弟姐妹。经过治疗的小鼠能够检测到大约45分贝的声音,这是正常谈话的水平,比未经治疗的小鼠安静16倍。听力保存最好的贝多芬老鼠能够听到25到30分贝的声音,与健康的老鼠几乎没有区别。
综上所述,这些发现表明,这种新型基因疗法有效地抑制了有缺陷的基因拷贝,并挽救了动物的听力,使其免于这种疾病典型的快速死亡。
由于这种疾病的特征是进行性听力损失,研究人员在几个月内评估了治疗对进展的影响。研究人员在小鼠出生后不久就对它们进行了治疗,并在长达六个月的时间里,每四周对接受治疗和未接受治疗的小鼠进行一次听力水平测试。在第一个月,未经处理的贝多芬小鼠可以听到低频声音,但在高频时听力明显下降。出生后6个月,未经治疗的贝多芬小鼠失去了全部听力。相比之下,经过治疗的贝多芬小鼠在低频下仍能保持接近正常的听力,有些甚至在高频下也能表现出接近正常的听力。
值得注意的是,没有携带基因缺陷的接受治疗的动物并没有因为基因治疗而经历任何听力损失——这一发现证明了该手术的安全性以及选择性靶向基因异常拷贝的能力。更令人鼓舞的是,一小部分接受治疗的贝多芬小鼠在接受了近一年的跟踪后,听力保持稳定,接近正常。
由于贝多芬缺陷的标志是内耳听力细胞的逐渐退化和死亡,研究人员使用电子显微镜来观察这些关键听力细胞的结构。正如预期的那样,在未经治疗的贝多芬小鼠中,研究人员发现听力细胞逐渐丧失,结构恶化。相比之下,治疗贝多芬小鼠和治疗健康小鼠老鼠两组听力细胞数量正常,结构完整或接近完整。
在最后的实验中,科学家们在携带贝多芬突变的人类细胞中测试了治疗的效果。DNA分析显示,治疗只对Tmc1基因的突变拷贝进行编辑,而对正常拷贝进行编辑。
由于其靶向单点基因突变的能力,这种方法有望治疗其他15种遗传性耳聋,这些耳聋也是由其他基因DNA序列中的单字母突变引起的听力基因.
此外,该团队表示,他们的技术可以适用于由单点突变引起的其他显性遗传疾病。为了确定它的假想效用,科学家们扫描了联邦ClinVar数据库——一个包含所有已知与人类疾病相关的基因突变的国家储存库。分析表明,基于该工具的特异性,它可以正确识别3759个缺陷基因变异,这些缺陷基因变异共同占显性人类的五分之一基因突变.
霍尔特说:“可以肯定的是,这只是漫长旅程中的第一步。“但我们在这里得到的是原则的证明,证明这种高度特异性、高度针对性的治疗方法可以被开发出来,选择性地抑制携带单点突变的基因,并有可能治疗许多其他形式的人类疾病。”
进一步探索