罕见神经退行性疾病的诊断和治疗进展

梅奥诊所的研究人员,以及国家和全球的合作者,已经开发出一种潜在的马查多-约瑟夫病,或脊髓小脑性共济失调3型(SCA3)的检测方法,这种疾病目前无法治愈。他们还阐明了与该疾病相关的基因靶点的作用。
这种遗传性疾病与ATXN3基因突变有关。这种影响中枢神经系统的突变出现在40 - 70岁之间,其特征是步态不稳,肌肉失去控制,运动和感觉神经衰退。症状可能类似于帕金森氏症或多发性硬化症。研究人员将他们的发现发表在科学转化医学.
在回顾性研究在美国,研究人员开始寻找一个分子靶点,以帮助评估SCA3及其所属的神经退行性疾病的潜在治疗方法。在这项工作中,指导研究人员的是梅奥诊所以前对卢伽雷氏病(也称为肌萎缩性侧索硬化症)的研究额颞叶痴呆C9orf72基因突变
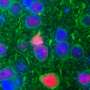
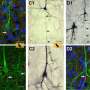
SCA3是由突变体的特征积累来定义的蛋白质: polyQ ataxin-3。研究人员认为,这种蛋白质在细胞内聚集,并通过多种方式干扰细胞活动而导致毒性。虽然目前还没有针对SCA3的治疗方法,但其目标是减少蛋白质积累。
该研究的第一作者Mercedes Prudencio博士说:“我们能够开发一种免疫分析或测试,可以量化人类生物液中突变蛋白的数量,如脑脊液和SCA3患者的血液。”“我们可以利用这项测试来确定旨在减少突变蛋白数量的新疗法的有效性。”普鲁登希奥博士是梅奥医学与科学学院神经科学助理教授。
该论文的资深作者之一Leonard Petrucelli博士说:“患者对SCA3的生物标志物的需求还没有得到满足,它可以告诉我们针对polyQ ATXN3蛋白的治疗是否有效。”“如果治疗能够更有针对性地减少突变蛋白,而不是正常蛋白,这将对该领域产生重大影响。”Petrucelli博士是Ralph B. and Ruth K. Abrams神经科学教授。
研究人员还澄清了一种名为a单核苷酸多态性在研究样本中,该基因与突变的ATXN3基因密切相关。
普鲁登西奥博士说:“虽然突变蛋白水平可以作为评估某些SCA3疗法是否有效的标志,但与突变蛋白密切相关的基因改变为开发专门针对它的疗法提供了新的机会。”该研究结果为SCA3疗法的加速临床试验铺平了道路。
从历史上看,SCA3在亚速尔群岛和葡萄牙人身上更常见。然而,在欧洲、亚洲和美洲都发现了这种疾病的病例。研究队列包括来自全球各地的患者样本,包括日本、墨西哥、荷兰、挪威、葡萄牙、瑞典和美国。伦敦大学学院附属共济失调中心多年来收集了来自欧洲国家的SCA3样本,验证了研究结果。
“这是团队科学领域真正的国际努力,”该论文的资深作者、医学博士兹比格涅夫·韦佐莱克(Zbigniew Wszolek)说。“SCA3是一种罕见而复杂的疾病,在这项工作上的合作对最终能够帮助正在遭受痛苦的患者和他们的家人有很大的帮助。”wzolek博士是梅奥医学与科学学院的神经学教授,以及神经退行性疾病的霍沃斯家族教授。
梅奥诊所的研究人员计划通过梅奥诊所共济失调项目跟踪SCA3患者,作为进一步研究这种神经退行性疾病的下一步。
进一步探索