研究人员确定了多囊性肾脏疾病的基因疗法靶标
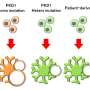
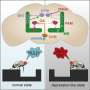
. Deleting the PKD1 miR-17 binding site markedly reduces cyst growth (right). Yellow indicates kidney collecting ducts and blue indicates nuclei. Credit: UT Southwestern Medical Center")
UT西南研究人员报道,通过删除microRNA的结合位点阻碍了常染色体显性多囊肾脏疾病(ADPKD)模型,阻止了肾脏囊肿的形成和生长,从而阻止了PKD1和PKD2基因表达的抑制作用。这些发现,发表在自然通讯,提出一种基因疗法的策略,具有阻止或治愈ADPKD的潜力。
“在25年以上,我们知道ADPKD是由PKD1或PKD2基因的突变引起的。然而,没有治疗策略可以追求这些根本原因,”该部门内科医学副教授Vishal Patel说。UTSW的肾脏科和该论文的对应作者。
ADPKD是最常见的人类遗传状况之一,也是肾衰竭最常见的遗传原因,影响了全球估计有1,250万人。ADPKD是一种遗传疾病,其中患者通常遗传一个突变的PKD1(或PKD2)和一份正常副本。
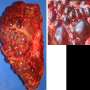
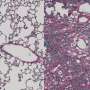
该疾病的特征是许多小液体囊性囊肿的经常形成,称为肾脏囊肿,当PKD1或PKD2的水平降至临界阈值以下时,它们被认为形成。当基因的正常拷贝无法产生足够的蛋白质polycystin-1/polycystin-2时,就会发生这种情况。
蛋白质是由基因的信使核糖核酸(mRNA)产生(或翻译)的。在mRNA链的一端是一个代码区域,有助于保护其免受降解,但也可以控制制造多少蛋白质。microRNA与mRNA代码区域的结合可以阻止翻译,从而导致蛋白质的产生。
PKD1包含a结合位点对于miR-17,在ADPKD模型中高度表达且活跃的microRNA。因此,帕特尔博士和他的同事询问阻止miR-17与PKD1的结合是否可以防止肾脏囊肿形成。
研究人员从PKD1 mRNA中删除了miR-17结合位点细胞培养和ADPKD鼠标模型。他们的结果表明,结合位点的缺失提高了mRNA链的稳定性,升高的多囊1水平以及肾脏囊肿生长的降低。此外,该小组发现,在囊肿形成后阻止miR-17与抗MIR-17药物结合的miR-17结合也降低了囊肿的生长,这表明这种相互作用可能是有希望的目标多囊肾(PKD)治疗。
帕特尔博士说:“在许多遗传条件下,一个因果基因的一个副本被突变,但另一个副本仍然是正常的。我们利用剩余正常副本的方法可能适用于除PKD以外的许多其他疾病。”