研究人员开发罕见纤毛病的基因疗法
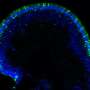
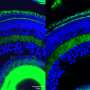
, NPHP5 (red), and rhodopsin (green). Below: Close-up view of organoid photoreceptor layer stained green for rhodopsin. Adapted from Kruczek et al, 2022. Credit: Anand Swaroop, Ph.D. and Kamil Kruczek, Ph.D., National Eye Institute")
美国国家眼科研究所(NEI)的研究人员开发了一种基因疗法,可以挽救一种莱伯氏先天性黑蒙(LCA)所影响的视网膜细胞的纤毛缺陷。LCA是一种导致儿童早期失明的疾病。利用患者来源的视网膜类器官(也称为视网膜培养皿),研究人员发现一种LCA是由视网膜的突变引起的NPHP5(也称为IQCB1)基因导致初级纤毛的严重缺陷,纤毛是一种几乎存在于身体所有细胞中的结构。这些发现不仅揭示了NPHP5蛋白在初级纤毛中的功能,而且也为治疗这种致盲疾病提供了潜在的治疗方法。NEI是美国国立卫生研究院的一部分。
“看到小孩子因为早期LCA而失明,真是太悲哀了。NPHP5缺乏在较轻的情况下会导致早期失明,在较严重的情况下,许多患者还会出现肾脏疾病视网膜变性该研究的首席研究员Anand Swaroop博士说,他是NEI神经生物学神经退行性变和修复实验室的高级研究员。“我们设计了一个基因治疗这种方法可以帮助防止患有这种疾病的儿童失明,通过进一步的研究,这种方法甚至可能有助于治疗这种疾病的其他影响。”
LCA是一种罕见的遗传病,它会导致眼睛后部感光视网膜的退化。至少25个不同基因的缺陷会导致LCA。虽然有一种基因疗法可以治疗一种类型的LCA,但其他所有类型的LCA都没有治疗方法。由突变引起的LCA的类型NPHP5相对罕见。在所有情况下,它都会导致失明,在很多情况下,它还会导致肾脏衰竭,这种情况被称为Senior-Lø肯综合症.
三位博士后Kamil Kruczek博士、Zepeng Qu博士和Emily Welby博士与研究团队的其他成员一起,从两名患有NPHP5在NIH临床中心这些干细胞样本被用来产生视网膜类器官,培养的组织簇具有许多实际的原生视网膜的结构和功能特征。患者衍生的视网膜类器官特别有价值,因为它们密切模仿实际患者的基因型和视网膜疾病表现,并为测试治疗干预提供了“类人”组织环境,包括基因疗法。与患者一样,这些视网膜类器官显示出光感受器的缺陷,包括失去部分光感受器叫做“外段”。
在健康的视网膜中,光感受器的外节含有视蛋白这种感光分子。当外部部分暴露在光线下时,光感受器就会启动一种神经信号,传递到大脑并调节视觉。光感受器的外段是一种特殊的初级纤毛这是一种古老的结构动物细胞.
在健康的眼睛中,NPHP5蛋白被认为位于初级眼基部的门状结构上纤毛这有助于过滤进入纤毛的蛋白质。先前对小鼠的研究表明NPHP5参与纤毛,但研究人员还不知道NPHP5在光感受器纤毛中的确切作用,也不清楚突变是如何影响蛋白质功能的。
在目前的研究中,研究人员发现在患者来源的视网膜类器官细胞中,NPHP5蛋白水平降低,另一种名为CEP-290的蛋白质水平也降低,CEP-290与NPHP5相互作用,形成初级纤毛门。(突变cep - 290构成了LCA最常见的病因。)此外,视网膜类器官的光感受器外节段完全缺失,视蛋白本应定位在外节段,却在光感受器细胞体的其他地方找到了。
当研究人员引入一种腺相关病毒(AAV)载体,其中包含功能版本的NPHP5作为基因治疗载体,视网膜类器官显示了视蛋白在外节段适当位置的明显恢复。研究结果还表明,功能性NPHP5可能稳定了初级纤毛门。
进一步探索