Gene therapy rapidly improves night vision in adults with congenital blindness
. DOI: 10.1016/j.isci.2022.105274")
Adults with a genetic form of childhood-onset blindness experienced striking recoveries of night vision within days of receiving an experimental gene therapy, according to researchers at the Scheie Eye Institute in the Perelman School of Medicine at the University of Pennsylvania.
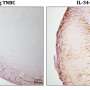
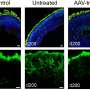
The病人had Leber Congenital Amaurosis (LCA), a congenital blindness caused by mutations in the gene GUCY2D. The researchers, whose findings are reported in the journaliScience, delivered AAV gene therapy, which carries the DNA of the healthy version of the gene, into the retina of one eye for each of the patients in accordance with the clinical trial protocol. Within days of being treated, each patient showed large increases, in the treated eye, of visual functions mediated by rod-type photoreceptor cells. Rod cells are extremely sensitive to light and account for most of the human capacity for low-light vision.
"These exciting results demonstrate that the basic molecular machinery of phototransduction remains largely intact in some cases of LCA, and thus can be amenable to gene therapy even after decades of blindness," said study lead author Samuel G. Jacobson, MD, Ph.D., a professor of Ophthalmology at Penn.
LCA is one of the most common congenital blindness conditions, affecting roughly one in 40,000 newborns. The degree of vision loss can vary from one LCA patient to another but all such patients have severe visual disability from the earliest months of life. There are more than two dozengenes的功能障碍可以导致LCA。
Up to 20% of LCA cases are caused by mutations in GUCY2D, a gene that encodes a key protein needed in retinal photoreceptor cells for the "phototransduction cascade"—the process that converts light to neuronal signals. Prior imaging studies have shown that patients with this form of LCA tend to have relatively preserved photoreceptor cells, especially in rod-rich areas, hinting that rod-based phototransduction could work again if functional GUCY2D were present. Early results with low doses of the gene therapy, reported last year, were consistent with this idea.
The researchers used higher doses of the gene therapy in two patients, a 19- year-old man and a 32-year-old woman, who had particularly severe rod-based visual deficits. In daylight, the patients had some, albeit greatly impaired, visual function, but at night they were effectively blind, with light sensitivity on the order of 10,000 to 100,000 times less than normal.
The researchers administered the therapy to just one eye in each patient, so the treated eye could be compared to the untreated eye to gauge treatment effects. The retinal surgery was performed by Allen C. Ho, MD, a professor of Ophthalmology at Thomas Jefferson University and Wills Eye Hospital. Tests revealed that, in both patients, the treated eyes became thousands of times more light-sensitive in low-light conditions, substantially correcting the original visual deficits. The researchers used, in all, nine complementary methods to measure the patients' light sensitivity and functional vision. These included a test of room navigation skills in low-light conditions and a test of involuntary pupil responses to light. The tests consistently showed major improvements in rod-based, low-light vision, and the patients also noted functional improvements in their everyday lives, such as "can [now] make out objects and people in the dark."
"Just as striking was the rapidity of the improvement following therapy. Within eight days, both patients were already showing measurable efficacy," said study co-author Artur V. Cideciyan, Ph.D., a research professor of Ophthalmology at Penn.
To the researchers, the results confirm that GUCY2Dgene therapyto restores rod-based photoreceptor functions—and suggest that GUCY2D–LCA patients with more severe rod-based dysfunction are likely to benefit most dramatically from the therapy. The practical message is that there should be an emphasis on rod vision measurements at screening of LCA candidates and in monitoring them throughout a treatment trial.
The findings, the researchers said, also underscore the remarkable fact that in some patients with severe congenital vision loss, the retinal cell networks that mediatevisionremain largely alive and intact, and need only the resupply of a missing protein to start working again, more or less immediately.