研究人员建立了新的模型来检查Usher综合征
. DOI: 10.1038/s41467-023-36431-1")
Usher综合征是一种罕见的遗传性疾病,是导致耳聋和失明的主要原因,其中2A型(USH2A)是最常见的形式。由USH2A基因突变引起的USH2A可包括出生时的听力丧失和进行性视力丧失,从而导致视网膜色素变性(RP)。RP会影响视网膜(眼睛的感光层),导致视网膜感光细胞的破坏,最初会导致夜盲症,随后逐渐丧失日常视力。目前没有针对USH2A的治疗方法。
此外,直到现在,还没有一项研究接近于研究这种疾病背后的实际机制——尽管建立一个反映患者表型的USH2A模型是未来治疗策略发展的一个重要目标。大多数Usher综合征模型成功地复制了听力损失观察到患者的视力问题,但未能建立模型。
在我们的研究中,发表于自然通讯“我们设计并生成了一个表达c.2299delG的模型,这是USH2A中最常见的人类疾病突变,”John S. Dunn生物医学工程教授Muna Naash报告说。
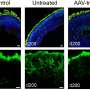
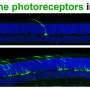
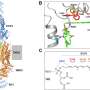
模型显示视网膜变性表达一种被截断的糖基化蛋白,这种蛋白错误地定位于感光细胞的内部片段。这种退化与视网膜功能下降、连接纤毛和外节的结构异常以及引导相互作用伙伴——长g蛋白受体1 (VLGR1)和旋转蛋白(WHRN)的错误定位有关。”
这些结果证明,实际突变蛋白的表达有利于再现USH2A视网膜表型,并为设计治疗干预策略提供了见解。
对模型视网膜的深入分析揭示了光感受器的结构异常,最终导致光感受器细胞死亡,导致视力丧失。由于该模型非常接近于这种疾病,它将允许Naash研究这种疾病的机制,并研究包括基因治疗在内的治疗方案。
“我们的模型显示视网膜变性与视网膜功能并持续支持有效的发展基因治疗治疗USH2A相关视力缺陷的平台,”纳什说。
纳什团队的其他成员包括摩尔生物医学工程教授穆阿亚德·r·乌拜迪;研究助理教授Lars Tebbe;Mustafa Makia和三个学生最近毕业:Maggie Mwoyosvi, Ryan Crane和Mashal Kakakhel。
更多信息:Lars Tebbe等人,引子突变c.2299delG导致其定位错误并破坏与whirlin和VLGR1的相互作用,自然通讯(2023)。DOI: 10.1038 / s41467 - 023 - 36431 - 1