研究发现板条疾病相关蛋白质生成新的溶酶体是至关重要的

神经元Ceroid Lipofuscinosis的毁灭性的神经退行性溶酶体储存障碍是一组在童年开始。CLN3基因的突变导致NCL叫板条疾病,特点是视力丧失的,运动,和认知。有针对性的有效的治疗方法不适用于这些障碍,因为大多数基因的生物角色负责这些疾病并不明确。
德克萨斯州贝勒医学院的研究人员和儿童医院在美国,电视节目遗传学和医学研究所和费德里科•二世大学在意大利已经发现CLN3是溶酶体的关键生物起源和自噬溶酶体改革(规律),发现一种新的疾病在板条疾病机制。
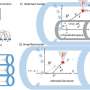
溶酶体作为细胞老化器和压实工具通过分解各种细胞的聚合物和碎片。溶酶体生物起源是一个关键的过程所需的新创(“从头开始”)代溶酶体规律是一个终端的自噬,细胞诱导在饥饿或其他压力条件下趋向下降的过程。
吞噬溶酶体期间消耗的初始步骤,在长期饥饿通过生成规律,一个过程的新功能溶酶体是由成熟的溶酶体。这里,研究小组发现CLN3新的溶酶体的生成至关重要,通过削弱新创溶酶体生物起源和规律通路,从而导致“岁”,溶酶体的积累也就无法正常运作。
该研究发表在自然通讯博士是由安德里亚·Ballabio贝勒大学教授、首席研究员Jan和丹邓肯神经研究所(邓肯新名词,贝勒大学助理教授和Alessia Calcagni博士。
“鉴于重要更新的溶酶体,自噬是细胞生存和功能,本研究具有极其深远的意义不仅对板条疾病但对许多其他条件是由缺陷引起的溶酶体贩运和存储、“Ballabio博士说。
CLN3通过高尔基氏复合体传输到溶酶体
缺乏一个可靠的工具来研究CLN3被研究者研究板条的障碍疾病,所以,小说生成的团队首先CLN3抗体。
令他们吃惊的是,他们发现CLN3不仅仅是溶酶体中被认为之前,但当第一次合成,它通过高尔基氏复合体传输获得一些修改在到达最终目的地之前,溶酶体。
CLN3导致损失扩大非功能性的溶酶体
接下来,探讨CLN3的生物作用(s),他们做了一个interactome分析识别所有的蛋白质,绑定到CLN3。这些实验表明,其合作伙伴是参与细胞内的蛋白质贩运途径和形成新的溶酶体,这表明它可能在这些过程起着重要的作用。
溶酶体蛋白质排序,cation-independent mannose-6-phosphate受体(CI-M6PR)被发现最丰富CLN3扶少团团员。大多数的溶酶体酶获得mannose-6-phosphate残留作为“地址标签”,然后被特定mannose-6-phosphate受体(类似于“发货人”)被路由到正确的endo-lysosomal隔间(“目的”)。
有趣的是,他们发现CLN3导致的损失mis-sorting CI-M6PR本身的溶酶体降解。正如所料,这扰乱了溶酶体生物起源。新的溶酶体显示全球消耗的几个降解酶。此外,缺乏CLN3导致溶酶体增大,聚合各种中间体参与自我吞噬,这是规律和溶酶体功能受损的一个特性。
溶酶体降解缺陷造成的mis-sorting CLN3-depleted CI-M6PR的细胞,废除最初消费溶酶体和他们的改革在饥饿,阻塞规律,因此,自噬。
CI-M6PR对CLN3-mediated至关重要规律
当研究人员在细胞中过表达CLN3缺乏CLN3基因,它导致溶酶体数量的大幅度增加,尤其是在长时间的饥饿,减少溶酶体的大小,增加了溶酶体酶的水平,促进了autophagy-dependent新的溶酶体的形成。相反,沉默CI-M6PR完全废除CLN3-mediated制管和溶酶体的改革。
“我们希望知道如何CLN3影响溶酶体的功能将帮助我们和其他人将来确定板条疾病有效的治疗方法,”Alessia Calcagni博士说。“此外,这项研究表明规律障碍和全球减少溶酶体的降解能力可能是一个可能的根本原因为其他ncl并将重要的测试在未来。”
更多信息:Alessia Calcagni et al,损失的板条疾病蛋白质CLN3导致mis-trafficking M6PR和缺陷autophagic-lysosomal改革,自然通讯(2023)。DOI: 10.1038 / s41467 - 023 - 39643 - 7